Queries can be made via textbox input or by file upload.
Query types:

By genomic position
Input should be in BED-like format, i.e.
chr pos pos ref alt
Genomic positions should be with reference to the GRCh37 assembly. Query entries (chr/pos/pos/ref/alt) should be tab-separated; ref and alt are optional; if ref and alt are not provided, first and second pos could be different and a range of results will be returned. Multiple queries should be separated by new line. Given the large size of the database, we limit the query size to 5k variants at a time.
By HGNC gene symbols
HGNC gene symbols should be separated by newline. Given the large size of the database, we limit the query size to 3 genes at a time.
By dbSNP ID
dbSNP IDs should be separated by newline. Given the large size of the database, we limit the query size to 5k variants at a time.
By VCF file
For the specification of a Variant Call Format (VCF) file, please visit here. Given the large size of the database, we limit the query size to 5k variants at a time, and 500 Mb for upload query (result will only be provided as downloadable file). The genome reference of the VCF file should be hg19/GRCh37.
Results
Results page for queries by genomic position/dbSNP/VCF
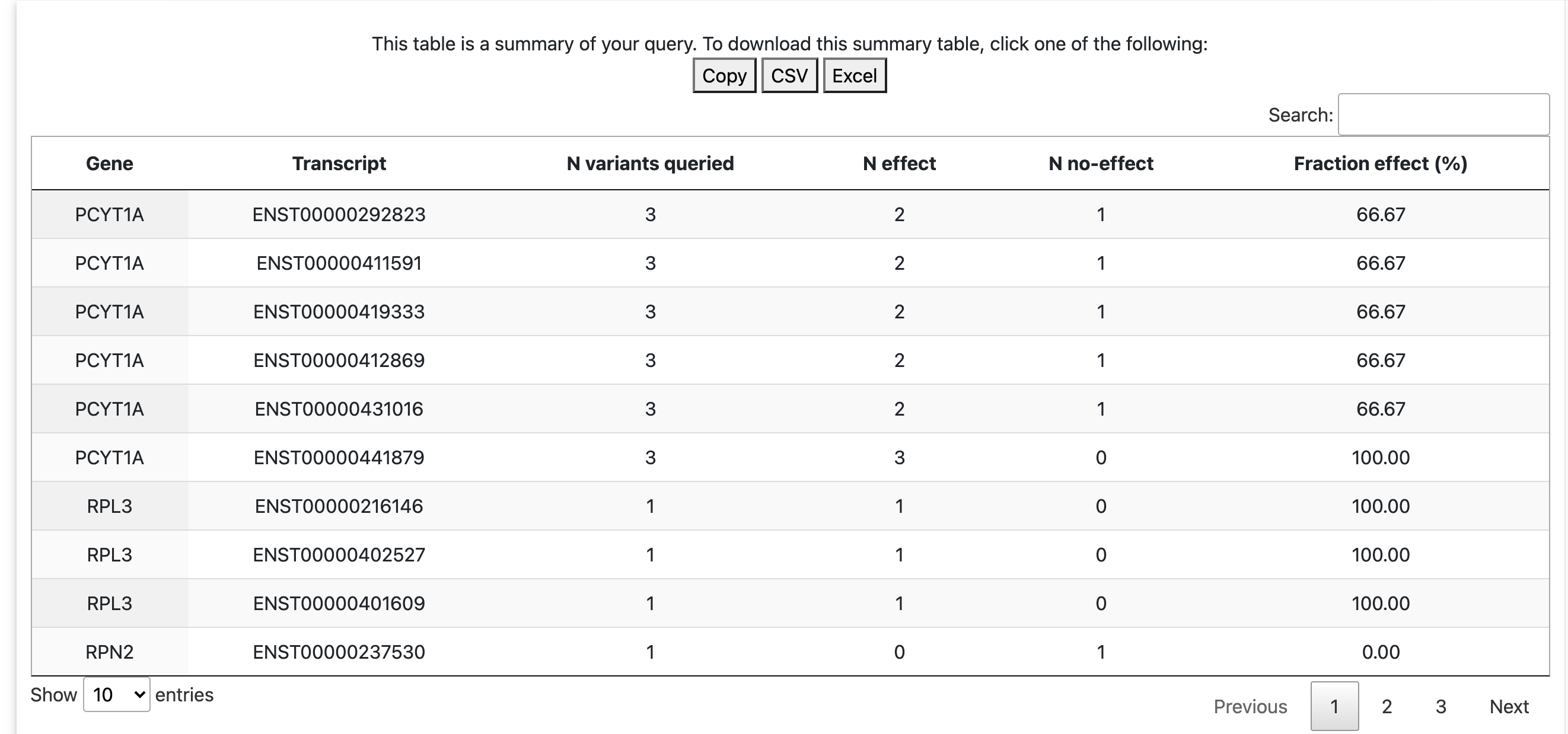
For the queries by genomic position/dbSNP/VCF, the result will have a summary table, indicating, for each Ensembl transcript, the number of variants queried and the number of predicted effect and no effect variants among them (and percentile of predicted effect variants). This summary table can be downloaded or copied.
There will also be a result table, where each row represents a query variant. Depending on the size of your query, it may take a while for the table to finish loading. The table can be sorted by a column by clicking the column header. Rows can be searched by key word via a search box in the upper right hand corner. The number of variants per page can also be adjusted. This table can be downloaded or copied.

There will also be a genomic map, which will allow you to zoom in and out to locate the positions of the queried variants in the human genome.

Results page for queries by gene symbol
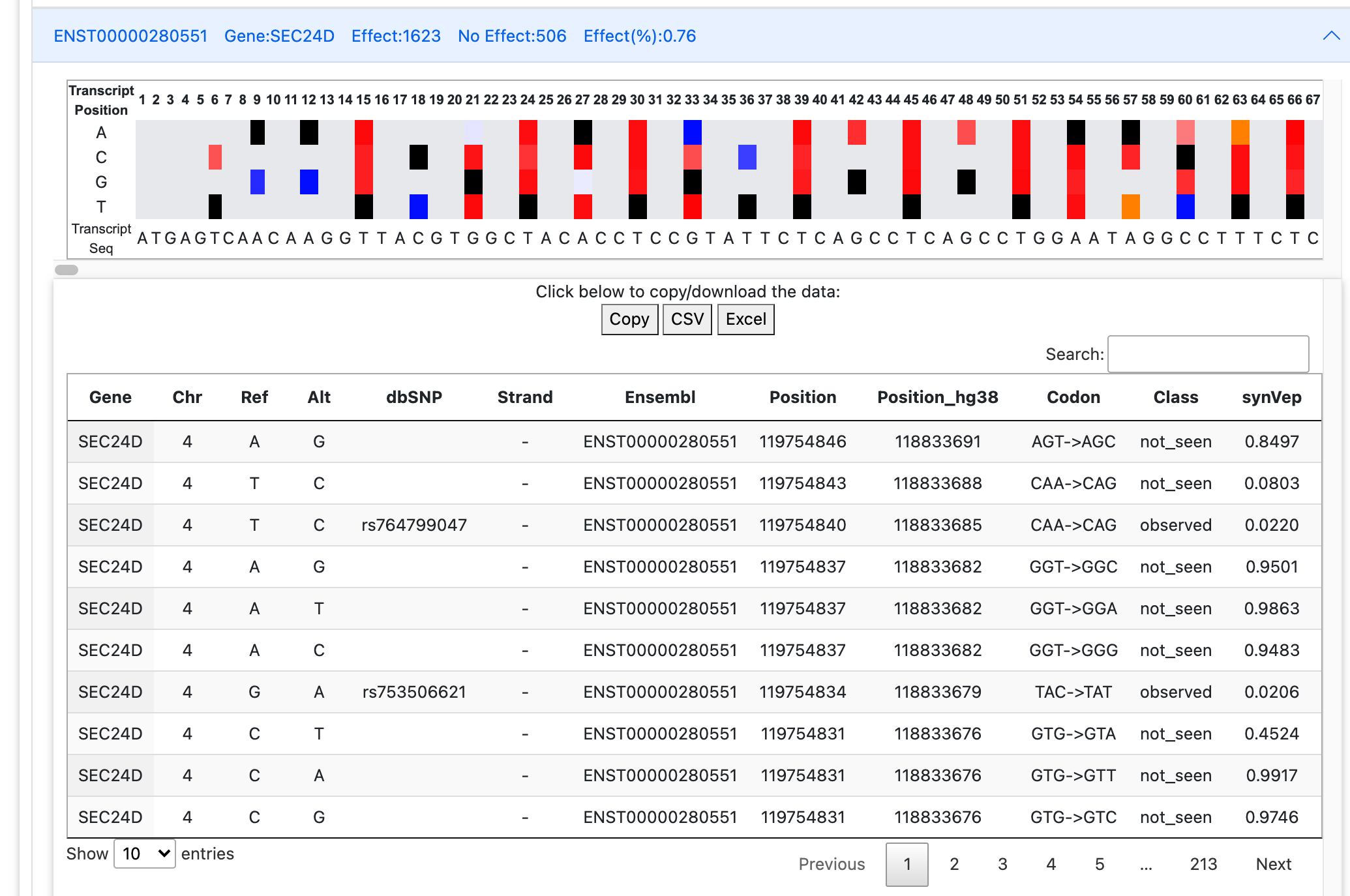
For queries by gene symbol, results are grouped by Ensembl transcript (see the expandable tabs). The expandable tabs summarize number of variants queried, number of predicted effect and no effect variants, and the percentile of predicted effect variants.
Underneath each expandable tab (i.e. one transcript), there will be a heatmap indicating the positions and
predictions for all variants possible within the transcript sequence:
top line of the heatmap is the nucleotide number;
bottom line of the heatmap is the nucleotide sequence;
black indicates the reference nucleotide at a position;
grey indicates that the variant is not synonymous;
red indicates variants with predicted effect. Color darkness corresponds to the synVep score (higher scores are darker);
blue indicates variants with no predicted effect. Color darkness corresponds to the synVep score (lower scores are darker);
orange highlights the "unobservable" class variants, where no predictions are available.
Below the heatmap is the variant table as described above. Note that, depending on the size of your query, it may take a while for the table to load.